FDA Issues New Enforcement Priorities for Unauthorized ENDS and Nicotine Pouch Products: What Manufacturers, Importers, and Retailers Should Know
FDA’s New Guidance Signals a More Targeted Enforcement Strategy for Vapes and Nicotine Pouches
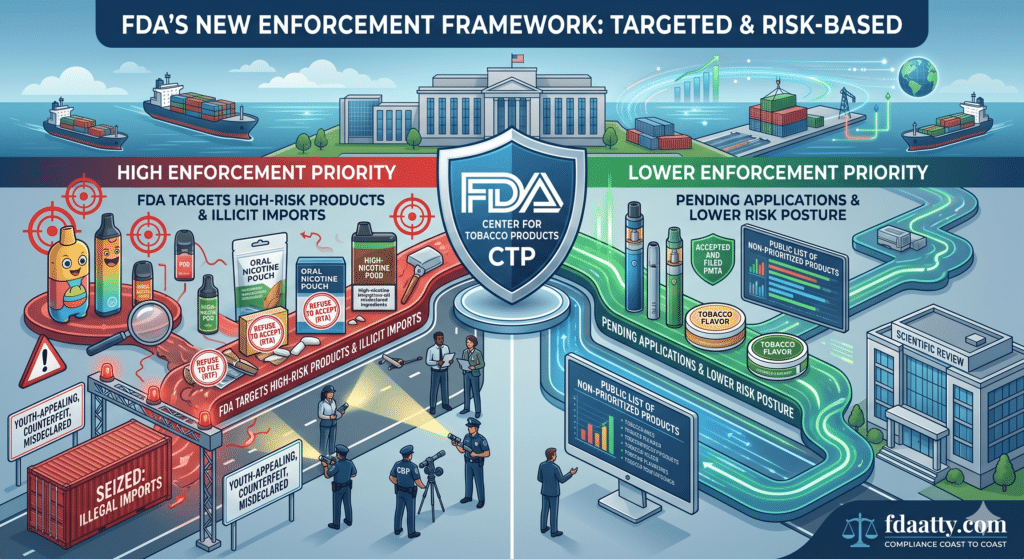
On May 8, 2026, the FDA issued final guidance titled “Enforcement Priorities for Certain New Tobacco Products Marketed Without Premarket Authorization.” The guidance applies to certain electronic nicotine delivery systems (ENDS) and oral nicotine pouch products that are currently marketed without FDA premarket authorization. FDA’s announcement emphasizes an “aggressive strategy” against illicit tobacco products, particularly illegal imports, counterfeit products, misdeclared products, and unauthorized ENDS and nicotine pouch products that have not undergone scientific review.
The guidance is important because it clarifies when the FDA does not intend to prioritize enforcement against certain unauthorized ENDS and nicotine pouch products—and, just as importantly, when the FDA may treat those products as enforcement priorities. For manufacturers, importers, distributors, and retailers, the guidance should not be read as permission to market unauthorized products. Rather, it is a risk-based enforcement policy that narrows the circumstances under which the FDA may exercise enforcement discretion.
In my practice advising ENDS manufacturers, importers, and retailers, the FDA’s shifting enforcement priorities have fundamentally changed our market entry strategies. I regularly counsel companies on the rigorous demands of the Premarket Tobacco Product Application (PMTA) process, as relying on a pending submission tracking number is no longer a viable shield from enforcement. Recently, I collaborated with a national distributor to map out a comprehensive, multi-phase testing strategy for their ENDS and nicotine pouch products—covering everything from aerosol HPHC chemistry and extractables/leachables to primary human behavioral studies. The reality is that the FDA expects robust, primary scientific data, and manufacturers must be prepared to invest in stability programs and in vitro toxicology if they want to survive Filing Review and stay on the shelves.
The Baseline Rule Has Not Changed: New Tobacco Products Require FDA Authorization
The Federal Food, Drug, and Cosmetic Act requires that new tobacco products may not be legally marketed without FDA premarket authorization. FDA states this directly in the guidance: all new tobacco products on the market without authorization are illegally marketed products.
For ENDS and nicotine pouch products, the relevant premarket pathway is generally a Premarket Tobacco Product Application (PMTA) or, where appropriate, a supplemental PMTA. FDA’s guidance explains that the agency generally does not intend to prioritize enforcement of the premarket authorization requirement where the product is subject to a pending application that has been accepted and filed, or is subject to certain supplemental PMTAs that have been accepted and pending for more than 180 days.
That distinction matters. A product with a PMTA merely “submitted” is not necessarily in the same enforcement posture as a product whose application has been accepted and filed and is in scientific review. The FDA specifically noted that many applications have been administratively incomplete and have received Refuse to Accept (RTA) or Refuse to File (RTF) determinations due to missing key information necessary for scientific review.
What Products May Fall Within FDA’s Lower Enforcement Priority Category?
FDA’s guidance applies to two categories of tobacco products:
- ENDS products, including e-liquids and devices that deliver aerosolized e-liquid when inhaled; and
- Oral nicotine pouch products, including products containing nicotine from any source.
For unauthorized ENDS and nicotine pouch products, the FDA generally does not intend to prioritize enforcement where the product is covered by a pending PMTA that has been accepted and filed, or a qualifying supplemental PMTA, subject to additional conditions. For non-tobacco-flavored ENDS products, the FDA states that the application must include data necessary to evaluate whether the product is appropriate for the protection of the public health.
This does not mean that FDA has authorized the product. It means only that, as a matter of enforcement priority, FDA may allocate enforcement resources elsewhere while the product remains under substantive review.
What Products Remain High Enforcement Priorities?
FDA’s guidance makes clear that products outside the described categories remain subject to prioritized enforcement. Even products that otherwise fall within the enforcement discretion policy may become enforcement priorities if they present youth-appealing or public-health concerns.
FDA identifies several factors that may trigger enforcement attention, including products that:
- depict cartoon-like fictional characters;
- disguise the product’s nature as a vaping product;
- resemble children’s toys, phones, or gaming platforms;
- contain high nicotine levels;
- are associated with serious or unexpected adverse experiences;
- lack child-resistant packaging required under the Child Nicotine Poisoning Prevention Act of 2015; or
- present a potential fire hazard.
FDA also emphasized that it will continue to coordinate with the Department of Justice, Customs and Border Protection, and other federal agencies to seize and destroy illegal tobacco products at the border.
For importers, this is a critical point. Products that are counterfeit, misdeclared, or imported without a defensible PMTA status may face significant border risk, including detention, refusal, seizure, or other enforcement action.
FDA Will Create a Public-Facing List of Products Not Prioritized for Enforcement
One of the most practical developments in the guidance is the FDA’s plan to create and maintain a public-facing webpage that identifies manufacturers and products the FDA generally does not intend to prioritize for enforcement under the policy.
FDA recommends that manufacturers with pending applications in scientific review contact the relevant regulatory health project manager (RHPM) if they wish to be included on the list. FDA also indicates that manufacturers may cross-reference relevant sections, modules, or pages of the pending PMTA rather than recreating information already included in the application.
This list could become highly significant for retailers, distributors, import brokers, investors, and counterparties conducting regulatory diligence. However, inclusion on the list should not be overstated. FDA cautions that providing the requested information does not guarantee that FDA will not pursue enforcement on a case-by-case basis, and that falling within the enforcement policy has no bearing on whether the product is likely to receive marketing authorization.
Why This Guidance Matters
The guidance reflects a practical reality: FDA does not have the resources to pursue every unauthorized tobacco product simultaneously. But FDA is also signaling a more targeted enforcement model focused on the products and actors it views as most problematic—particularly illicit imports, counterfeit products, misdeclared products, youth-appealing products, and products that lack a serious, reviewable PMTA package.
For industry, the regulatory message is straightforward: the quality and procedural status of the PMTA matters. A pending application that has been accepted and filed is materially different from an incomplete submission that has not survived the FDA’s initial review screens. Companies should not assume that a filing, receipt confirmation, or prior correspondence with the FDA is enough to reduce enforcement risk.
Practical Steps for Manufacturers, Importers, and Retailers
Companies operating in the ENDS or nicotine pouch space should consider taking the following steps:
Confirm PMTA status. Determine whether each marketed SKU is covered by a PMTA or supplemental PMTA that has been accepted and filed, or is otherwise in a posture described by the guidance.
Audit product identifiers. FDA specifically noted deficiencies such as incomplete tobacco product identification, manufacturing information, test method validation, ingredient information, HPHC data, and device operation data. Companies should ensure that product records, application cross-references, labels, and import documentation are internally consistent.
Evaluate youth-appealing features. Packaging, character imagery, product shape, flavor presentation, and digital marketing should be reviewed carefully. Products resembling toys, phones, gaming devices, or otherwise youth-oriented formats remain high risk.
Review nicotine content and safety signals. High nicotine levels, adverse experience data, battery or fire risks, and child-resistant packaging deficiencies may materially alter enforcement risk.
Prepare for public-list diligence. Manufacturers with pending applications in scientific review should consider whether to seek inclusion on FDA’s planned public-facing list, while understanding that listing does not equal authorization.
Strengthen import controls. Importers should evaluate customs descriptions, tariff classifications, manufacturer identification, product authorization status, and documentation provided to brokers. Misdeclared or counterfeit products are squarely within FDA’s stated enforcement focus.
Key Takeaway
FDA’s May 2026 guidance does not legalize unauthorized ENDS or nicotine pouch products. Instead, it creates a more transparent framework for how FDA intends to allocate enforcement resources while PMTAs and supplemental PMTAs are pending. Companies that have complete, accepted, and filed applications—and that avoid youth-appealing or elevated safety-risk features—may be in a comparatively lower enforcement priority category. Companies without a defensible PMTA posture, or with products that raise youth appeal, safety, counterfeit, import, or misdeclaration concerns, should expect continued and potentially aggressive FDA scrutiny.
For manufacturers, importers, distributors, and retailers, this is the time to conduct a product-by-product regulatory status review and ensure that PMTA records, import documentation, labeling, packaging, and mar

Are you in trouble with the FDA?
Don’t panic — you’ve got backup. Download 5 Tips to Help You Navigate FDA Enforcement and learn how to resolve the situation right now.